JMS Pearce
Hull, England

In the history of medicine, few writers can have received a finer accolade than that bestowed by William Osler on George Huntington. Osler commented: “In the whole range of descriptive nosology there is not to my knowledge, an instance in which a disease has been so accurately and fully delineated in so few words.”1 He was referring to Huntington’s account of hereditary chorea, often cited as the classical example of autosomal dominant inheritance. Its discovery and subsequent case reports are studded with both mystery and drama2—the mystery of the nature of the disease, the drama of its clinical features, and its inexorable progressive plundering of mind and body.
In early May 1633, the ship Elizabeth Bonaventure with John Graves, its Master, and ninety-five passengers set sail from Yarmouth in Norfolk England, arriving in Boston, Massachusetts on June 15. One of its passengers, Simon Huntington, Sr., died of smallpox during the voyage. His wife Margaret and four children, including Simon, Jr. (1629–1706) aged four, settled in Norwich, New London, Connecticut.
Simon Huntington, Jr.’s great-great-great grandson Abel (1777–1858) became an esteemed doctor who moved to East Hampton, Long Island, New York. Two medical generations later, Abel’s grandson George Huntington (1850–1916) was born in East Hampton and trained with his father, Dr. George Lee Huntington, in his practice. He attended the College of Physicians and Surgeons at Columbia University, graduating in 1871–2.3 George’s MD thesis on Sydenham’s chorea included in the last section cases of a type of chorea he believed to be present “exclusively on the east end of Long Island.” In addition to the chaotic involuntary movements of chorea, he stressed two distinctive features. Firstly, the familial occurrence: “an heirloom from generations away back in the dim past,” and secondly, “as the disease progresses the mind becomes more or less impaired, in many amounting to insanity, until death relieves them of their sufferings.”
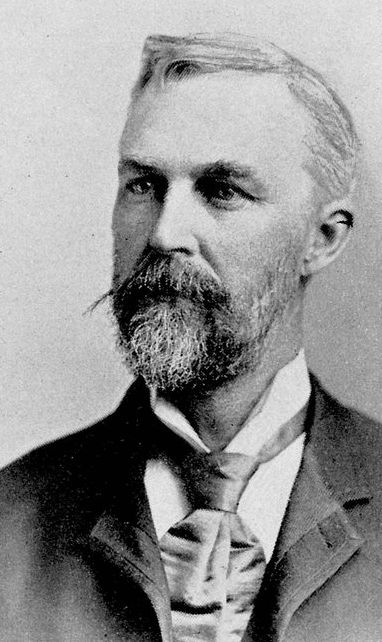
{kind=link}
Shortly after graduation George Huntington moved to Pomeroy, Ohio, where he set up his own practice. He had first encountered the disease when visiting patients with his father. He was twenty-two years old when on February 15, 1872 he read his paper on chorea to the Meigs and Mason Academy of Medicine in Middleport, Ohio.
And now I wish to draw your attention more particularly to a form of the disease which exists, so far as I know, almost exclusively on the east end of Long Island.…The hereditary chorea, as I shall call it, is confined to certain and fortunately a few families, and has been transmitted to them, an heirloom from generations away back in the dim past. It is spoken of by those in whose veins the seeds of the disease are known to exist, with a kind of horror, and not at all alluded to except through dire necessity, when it is mentioned as “that disorder.” It is attended generally by all the symptoms of common chorea, only in an aggravated degree, hardly ever manifesting itself until adult or middle life, and then coming on gradually but surely, increasing by degrees, and often occupying years in its development, until the hapless sufferer is but a quivering wreck of his former self.
It is as common and is indeed, I believe, more common among men than women, while I am not aware that season or complexion has any influence in the matter. There are three marked peculiarities in this disease:
1. Its hereditary nature… one or more of the offspring almost invariably suffer from the disease, if they live to adult age. But if by any chance these children go through life without it, the thread is broken and the grandchildren and great-grandchildren of the original shakers may rest assured that they are free from the disease.
2. A tendency to insanity and suicide… the mind becomes more or less impaired, in many amounting to insanity… until death relieves them of their sufferings.
3. Its manifesting itself as a grave disease only in adult life… I do not know of a single case that has shown any marked signs of chorea before the age of thirty or forty years.4
The patients described were from his father’s and grandfather’s practices.5 Addressing the New York neurological society in 1909, he recalled:
…riding with my father on his professional rounds, I saw my first case of “that disorder” which was the way the natives always referred to the dreaded disease . . . we came upon two women, mother and daughter, both tall, thin, almost cadaverous, both bowing, twisting, grimacing . . . I stared in wonderment, almost fear.6
An earlier mention of the disorder was given by John Elliotson in The Lancet in 1832. A letter by Reverend CO Waters in the first edition of Robley Dunglison’s Practice of Medicine in 1842 described in a group of residents in New York State “a form of chorea, vulgarly called magrums.” This included a description of the chorea, its progression, and its hereditary trait. Gorman reported similar instances in Philadelphia in the 1848 edition. The Norwegian Johan Christian Lund in 1860 also described typical choreic symptoms of a hereditary nature, but his paper was not translated into English until 1959.7
Huntington’s affected patients were further investigated. In 1923 Jelliffe and Frederick Tilney analyzed the history of HD sufferers in New England. And in 1932 a Connecticut psychiatrist, Percy R Vessie, traced twelve generations allegedly descended from three young men and their wives who left Bures in Essex for Suffolk bound for Salem, Massachusetts in 1630. Over a thousand descendants were thought to have succumbed, many being tried for witchcraft since their chorea was regarded as “a derisive pantomime of the sufferings of the Saviour during crucifixion.”8 Vessie seemed to accept mistakenly that accusations of witchcraft in women and misconduct of the men indicated the likely presence of the disease.9 His work has been subsequently described as flawed. Huntington’s chorea has been propagated largely, but not exclusively, through the descendants of several colonial families.10
A less common juvenile variant was later identified, usually inherited from an affected father. In addition to cognitive changes and chorea, limb rigidity, hypokinesia, and seizures figure prominently. In 1908 the New York neurologist William Browning devoted an entire issue of his journal Neurographs to Huntington’s chorea, emphasizing the worldwide interest in the disease.5
In 1874, Huntington moved to New York, practicing in Duchess County. He retired in 1915 and died a year later of pneumonia. He was regarded as an amusing, modest, kindly family man, keen on natural history, drawing, and music who claimed no expertise in research. His only other published paper was his recollections, given to the New York Neurological Society in 1910.6
A single dominant gene encoding a toxic protein causes Huntington’s disease (HD). Its pathology lies in a progressive, selective neural cell loss and atrophy mainly in the caudate nucleus and putamen with atrophy of the cerebral white matter.
It is typically inherited from an affected parent, who carries a mutation in the “huntingtin” gene (HTT). In 1983 the huntingtin gene was found using DNA markers.11 The huntingtin protein results from an expanded CAG repeat leading to an expanded toxic polyglutamine.12 HD genetic testing is now possible for diagnosis. Presymptomatic and prenatal testing can determine the carrier status, but considerable ethical and emotional consequences confound their general use. Treatment is symptomatic, and so far reducing huntingtin production by treatment (with tominersen, an antisense oligonucleotide designed to block the instructions cells use to make toxic proteins) in humans has been unsuccessful.
The disease is fatal with a life expectancy of about fifteen to thirty years after diagnosis. After Leonore Wexler was diagnosed with HD in 1968, her daughters Alice, a historian, and Nancy, a neuropsychologist, with their father Milton Wexler created the Hereditary Disease Foundation,13 which has spawned much valued genetic research, and attempts to find effective treatments.
References
- Osler W. Remarks on the varieties of chronic chorea, and a report upon two families of the hereditary form, with one autopsy. The Journal of Nervous and Mental Disease 1893; 20(2):97.
- Dahlkamp J. Incurably curious: mystery and drama in clinical case reports. Hektoen International Spring 2014.
- Critchley M. The history of Huntington’s chorea. Psychological Medicine 1984; 14:725-727.
- Huntington G. On chorea. Med Surg Rep 1872; 26:317-21. Full text available at: https://en.wikisource.org/wiki/On_Chorea.
- Wexler A, Wild EJ, Tabrizi SJ. George Huntington: a legacy of inquiry, empathy and hope. Brain 2016; 139(Pt 8):2326-33.
- Huntington G. Recollections of Huntington’s chorea, as I saw it at East Hampton, Long Island, during my boyhood. J Nerv Ment Dis 1910; 37(4):255–257.
- Harper B. Huntington disease. J R Soc Med 2005; 98(12):550.
- Vessie P Nerv Ment Dis 1932;76:553-73. Cited by RN Dejong in: Haymaker’s Founders of Neurology, 2nd ed 1970; 452-5.
- Wexler A. Stigma, history, and Huntington’s disease. Lancet 2010; 3;376(9734):18-9
- Hans MB. Gilmore TH. Huntington’s chorea and genealogical credibility. Journal of Nervous and Mental Disease 1969; 148(1):5-13.
- Gusella JF, Lee JM, MacDonald ME. Huntington’s disease: Nearly four decades of human molecular genetics. Hum Mol Genet 2021; 30(R2):R254-63.
- La Spada AR, Roling DB, Harding AE, et al. Meiotic stability and genotype–phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nature Genetics 1992; 2(4):301-4.
- Wexler NS. Huntington’s Disease: Advocacy Driving Science. Annual Review of Medicine 2012; 63:1, 1-22.
JMS PEARCE is a retired neurologist and author with a particular interest in the history of medicine and science.
Highlighted in Frontispiece Volume 15, Issue 2 – Spring 2023