

Volume 16, Issue 2 – Spring 2024 ISSN 2155-3017
The best of Charles Dickens
The city of Padua
| The revolution of Andreas Vesalius, Fabio Zampieri, Alberto Zanatta The origin and evolution of Padua hospitals, Alberto Zanatta, Fabio Zampieri University of Padua School of Medicine, JMS Pearce Beginnings of bedside teaching in Padua: Montanus Realdo Colombo (ca. 1515–1559) Adrianus Spigelius, the last great Paduan anatomist |
Women pioneers
| Elizabeth Casson, Victoria Bates Women in medicine in Serbia, Bojana Cokić Ida Sophia Scudder, Angela Joseph Alexa Canady: First black woman neurosurgeon, Howard Fischer |
Hektorama Feature: United States Hospitals
 Michael Reese Hospital
Michael Reese Hospital Bellevue Hospitals
Bellevue Hospitals Freedmen’s Hospital Washington D.C.
Freedmen’s Hospital Washington D.C.  Johns Hopkins Hospital
Johns Hopkins Hospital Maynard-Columbus Hospital, Nome, Alaska
Maynard-Columbus Hospital, Nome, Alaska Provident Hospital, Chicago
Provident Hospital, Chicago Rush Medical College, Chicago
Rush Medical College, Chicago Roosevelt Hospital, New York
Roosevelt Hospital, New York Old Cook County Hospital
Old Cook County Hospital Walter Reed Army Medical Center
Walter Reed Army Medical Center Harborview Hospital, Seattle
Harborview Hospital, Seattle San Francisco General Hospital
San Francisco General Hospital Massachusetts General Hospital
Massachusetts General Hospital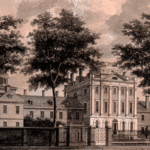 Pennsylvania Hospital
Pennsylvania Hospital Cincinnati Children’s Hospital
Cincinnati Children’s Hospital Peter Bent Brigham Hospital
Peter Bent Brigham Hospital Boston Dispensary
Boston Dispensary Williamsburg Hospital, Virginia
Williamsburg Hospital, Virginia St. Vincent Hospital Indianapolis
St. Vincent Hospital Indianapolis Alcatraz Hospital
Alcatraz Hospital Ellis Island Hospital
Ellis Island Hospital Joslin Diabetes Center
Joslin Diabetes Center Montefiore Medical Center, NY
Montefiore Medical Center, NY The Illinois Eye and Ear Infirmary
The Illinois Eye and Ear Infirmary Hillman Hospital, Birmingham, Alabama
Hillman Hospital, Birmingham, Alabama
Our general Grand Prix competition is currently closed. Follow us for news and announcements!